Исследователи из Центра вычислительной физики МФТИ разработали автоматический метод, который позволит объективно определять момент, когда система действительно достигает равновесия в молекулярно-динамической симуляции. Вместо привычного «на глаз»-анализа они предложили точный и воспроизводимый подход, критически важный для надежного моделирования сложных молекулярных систем. Статья опубликована в журнале Molecular Simulation.
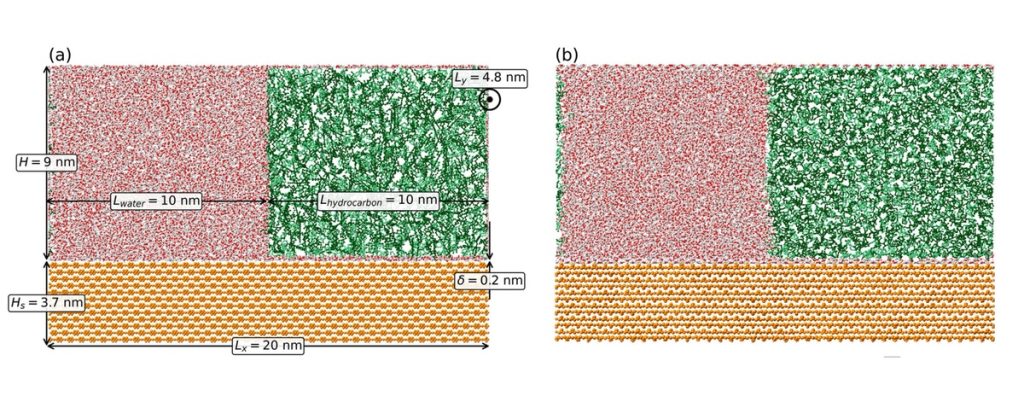
Определение точки равновесия в молекулярно-динамических симуляциях по границе двух несмешивающихся жидкостей, находящихся внутри узких нанопор,— одна из ключевых задач современной вычислительной физики. В таких системах, например углеводород—вода—кальцит в нанопоре, разные параметры — контактный угол, толщина смачивающего слоя и распределение плотности внутри капли — приходят к равновесию на разных временных шкалах. Поэтому видимая стабилизация формы капли не означает, что вся система находится в равновесии. Это приводит к ошибкам при усреднении данных и неверным выводам о смачиваемости, что критично для моделирования процессов добычи нефти.
«У нас периодически возникает потребность в проведении большого количества молекулярно-динамических расчетов. Необходимо как-то определять точку выхода на равновесие того или иного физического параметра системы для повышения качества данных»,— объяснил Тимур Гуськов, младший научный сотрудник Центра вычислительной физики МФТИ, аспирант кафедры вычислительной физики конденсированного состояния и живых систем МФТИ.
Авторы впервые применили современный алгоритм автоматического усечения данных на основе статистического метода к интерфейсам (граница между жидкостями) жидкость—жидкость в нанопоре кальцита, на примере чистый н-декан, чистый бензол и их смесь. Для прямой проверки плотности внутри капли они разработали новый дескриптор MF — математический показатель, характеризующий профиль плотности капли. Он не привязан к геометрии интерфейса, что позволяет отслеживать равновесие отдельных компонент даже в смесях.
Расчеты показали, что в системах углеводород—вода—кальцит контактный угол и макроскопическая форма капли стабилизируются в 10–200 раз быстрее, чем ее внутренняя структура. Перераспределение молекул углеводородов внутри капли может продолжаться еще десятки наносекунд после того, как форма капли уже «замерла».
Также они обнаружили, что при слабом усреднении метод PANDA-NN (автоматический расчет контактного угла по плотностным профилям) систематически завышает угол до +4,7°. Поэтому ученые предложили практический протокол: сначала определять момент равновесия при малом усреднении, а затем пересчитывать угол при большем усреднении. Это устраняет систематическую ошибку и повышает точность расчета.
«Плотность устаканивается дольше, чем форма интерфейса. То есть для понимания структуры капли необходимо моделировать систему несколько дольше, чем для обычных расчетов контактных углов. В особенности это важно для систем, в которых диффузия и адсорбция происходят медленно»,— подчеркнул Илья Копаничук, старший научный сотрудник Центра вычислительной физики МФТИ.
Предложенный подход делает результаты молекулярного моделирования надежными и воспроизводимыми. Он особенно востребован в нефтедобыче (карбонатные коллекторы), хранении CO₂, разработке мембран и наноматериалов.
«Впоследствии мы планируем изучать динамические свойства ограниченных интерфейсов, а также влияние шероховатости поверхностей на их смачиваемость. В этом нам помогут проработанные в данном исследовании подходы, которые уже активно внедряются в наши расчеты»,— поделился Николай Кондратюк, исполнительный директор Центра вычислительной физики МФТИ.
Работа выполнена при поддержке Российского научного фонда (грант № 25-13-00313).
Научная статья: Guskov T., Gerke K., Kondratyuk N., Khlyupin A. & Kopanichuk I. 2026. Assessing density equilibration in confined Liquid–Liquid interfaces. Molecular Simulation, p. 1–10. https://doi.org/10.1080/08927022.2026.2650110